Sindromul Kallmann
Astrid Leitner a studiat medicina veterinară la Viena. După zece ani de practică veterinară și nașterea fiicei sale, ea a trecut - mai întâmplător - la jurnalism medical. A devenit rapid clar că interesul ei pentru subiectele medicale și dragostea ei pentru scris erau combinația perfectă pentru ea. Astrid Leitner locuiește cu fiica, câinele și pisica în Viena și Austria Superioară.
Mai multe despre experții Tot conținutul este verificat de jurnaliștii medicali.Sindromul Kallmann este o boală congenitală în care sunt produși hormoni sexuali puțini sau deloc. Cei afectați nu trec prin pubertate; dacă nu sunt tratați, bărbații și femeile sunt sterili. În plus, simțul mirosului este de obicei deranjat: pacienții cu KS miros doar foarte limitat sau deloc. Citiți aici tot ce trebuie să știți despre „sindromul olfactogenital”!
Coduri ICD pentru această boală: codurile ICD sunt coduri recunoscute la nivel internațional pentru diagnostice medicale. Acestea pot fi găsite, de exemplu, în scrisorile medicului sau pe certificatele de incapacitate de muncă. E23

Prezentare scurta
- Ce este sindromul Kallmann? Tulburare congenitala de dezvoltare care duce la lipsa hormonilor sexuali si astfel la absenta pubertatii. În plus, majorității pacienților le lipsește simțul mirosului.
- Cauze: Modificări genetice congenitale (mutații)
- Factori de risc: Boala apare în familii la aproximativ 30% dintre pacienți.
- Simptome: Lipsa dezvoltării pubertății (subdezvoltarea penisului, a testiculelor și a prostatei, puțul pubian, axila și părul corpului, lipsa creșterii bărbii, absența primei perioade menstruale), infertilitatea, lipsa de plăcere, simțul mirosului este absent sau foarte mare consecință redusă, pe termen lung: osteoporoză
- Diagnostic: Simptome fizice precum lipsa dezvoltării caracteristicilor sexuale secundare, analiza hormonală, test genetic, ultrasunete, tomografie cu rezonanță magnetică, tomografie computerizată, radiografie
- Tratament: terapie de substituție cu medicamente hormonale
- Prevenire: Nu este posibilă prevenirea
Ce este sindromul Kallmann?
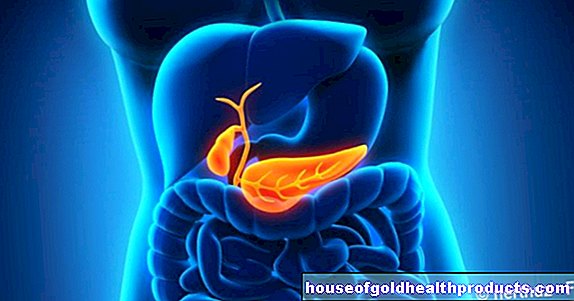
Sindromul Kallmann (KS, sindromul olfactogenital, sindromul De-Morsier-Kallmann) este o tulburare congenitală de dezvoltare a creierului. Se asigură că cei afectați nu produc hormoni sexuali și, prin urmare, nu apare maturitatea sexuală. În plus, majoritatea pacienților cu SC suferă de anosmie, ceea ce înseamnă că nu percep mirosuri.
Cauza bolii este o modificare genetică (mutație) care afectează deja dezvoltarea embrionară în uter. În centrul de control hormonal al creierului (hipotalamus) lipsesc anumite celule care sunt responsabile pentru controlul suprem al producției de hormoni sexuali în viața ulterioară. În plus, modificarea genei înseamnă că centrul olfactiv din creier (bulb olfactiv) nu este sau este doar incomplet dezvoltat. Persoanele afectate nu miros nimic sau doar într-o măsură foarte limitată.
Boala poartă numele psihiatrului german Franz Josef Kallmann, care a descris-o în 1944 drept „hipogonadism (lipsa hormonilor sexuali) și anosmie (lipsa mirosului)”. Termenul sinonim „Sindromul De Morsier-Kallmann” se referă și la neurologul elvețian Georges de Morsier, care a efectuat și studii asupra sindromului Kallmann.
Hipogonadism secundar
Sindromul Kallmann este o sub-formă a hipogonadismului secundar. Aceasta înseamnă o lipsă de hormoni sexuali, care este declanșată de o defecțiune în centrul de control hormonal din creier (glanda pituitară). Gonadele (ovarele, testiculele) funcționează normal, dar primesc semnale mici sau deloc despre producerea hormonilor sexuali. Dacă există animie (lipsa mirosului) pe lângă deficitul hormonal, medicii vorbesc despre sindromul Kallmann.
frecvență
Sindromul Kallmann este o boală rară; bărbații sunt mai frecvent afectați decât femeile: în medie, în jur de unul din 10.000 de bărbați și unul din 50.000 de femei îl suferă.
Simptome
Principalele simptome ale sindromului Kallmann sunt simțul mirosului foarte redus sau absent și absența dezvoltării pubertății. Cât de severe sunt simptomele variază de la pacient la pacient. Dacă nu sunt tratate, ambele sexe rămân sterile.
Lipsa pubertății: hormonii sexuali sunt esențiali pentru debutul pubertății. Dacă sunt prezenți prea puțini sau deloc hormoni sexuali, pubertatea și, prin urmare, dezvoltarea caracteristicilor sexuale secundare nu va avea loc.
Simptome la băieți:
- Păr pubian, axilar și corporal puțin sau deloc
- Barba lipsă
- Eșecul de a-și rupe vocea
- Micropenis și testicule mici: la bărbații tineri, penisul și testiculele nu se dezvoltă.
- Testicule nedescendente (adesea evidente la copilărie)
- Statura înaltă: la persoanele sănătoase, plăcile de creștere din oasele lungi se închid de îndată ce se atinge un anumit nivel de hormon sexual. Dacă hormonii lipsesc, cei afectați cresc disproporționat. Sunt mai înalți decât părinții lor și au brațe și picioare lungi.
Simptome la fete:
La fete, simptomele sunt de obicei mai puțin pronunțate. Adesea singurul simptom este absența primei menstruații (amenoreea primară), motiv pentru care boala este adesea recunoscută târziu. Spre deosebire de băieți, dezvoltarea fizică este în mare măsură normală în ciuda bolii. De exemplu, sânii sunt de obicei dezvoltați normal.
Simptome la bărbați adulți:
Bărbații cu sindrom Kallmann au de obicei densitate osoasă redusă (osteoporoză) și masă musculară. Adesea au un aspect feminin, extern datorită unei distribuții feminine a grăsimilor. Aveți disfuncție erectilă și sunteți steril.
Scăderea sau absența simțului mirosului: pacienții cu KS nu percep mirosurile (anosmie) sau percep doar mirosurile slab (hiposmie). Acest simptom trece adesea neobservat deoarece oamenii sunt obișnuiți să nu miroasă nimic de la naștere. Lipsa simțului mirosului poate duce la situații periculoase în cazuri individuale, de exemplu dacă cei afectați nu percep mirosul de arsură sau mirosul alimentelor alterate.
Malformații: Alte malformații fizice apar mai rar la pacienții cu SK. Acestea sunt, de exemplu, tulburări de auz, buză despicată, maxilar și palat sau dinți lipsă. Unii oameni se nasc cu un singur rinichi. Dezvoltarea mentală este de obicei normală în KS.
Osteoporoza: hormonii sexuali precum testosteronul și estrogenul joacă un rol important în mineralizarea osoasă. Dacă lipsesc hormonii, oasele nu sunt la fel de stabile ca la persoanele sănătoase - crește riscul fracturilor osoase.
Cauze și factori de risc
Cauza sindromului Kallmann este o schimbare genetică înnăscută (mutație).De obicei, apare spontan, în aproximativ 30 la sută din cazuri este transmisă descendenților de la unul sau de la ambii părinți.
Modificarea genetică afectează deja dezvoltarea embrionară în uter: celulele pentru simțul mirosului și cele responsabile de controlul gonadelor (ovare, testicule) provin din celulele progenitoare comune.
În sindromul Kallmann, dezvoltarea acestor celule progenitoare este perturbată de schimbarea genetică. Ca urmare, celulele care sunt responsabile pentru controlul suprem al producției de hormoni sexuali și celulele olfactive sunt insuficient dezvoltate. Persoanele afectate nu dezvoltă pubertate și nu percep mirosuri.
Hormoni sexuali
Hipotalamusul (o regiune din diencefal) este centrul de control hormonal al organismului. La persoanele sănătoase, hipotalamusul eliberează hormonul GnRH (hormon care eliberează gonadotropina). La rândul său, aceasta stimulează glanda pituitară să elibereze hormonii LH (hormonul luteinizant) și FSH (hormonul foliculostimulant).
LH și FSH acționează asupra gonadelor (ovare, testicule), care produc în cele din urmă hormonii sexuali: la femei, hormonii sexuali feminini estrogen și progesteron promovează maturarea celulelor ouă și declanșează ovulația, la bărbați hormonul testosteron provoacă formarea de spermă.
În timpul pubertății începe producția de hormoni sexuali și odată cu aceasta maturizarea sexuală. Dacă, la fel ca în sindromul Kallmann, este puțin sau deloc prezent GnRH, sunt produși prea puțini sau deloc hormoni sexuali și pubertatea nu se dezvoltă. Dacă nu sunt tratați, cei afectați nu experimentează maturitatea sexuală și rămân sterili.
Factori de risc
În majoritatea cazurilor, schimbarea genetică are loc spontan. Nu este clar cum se întâmplă acest lucru. În aproximativ 30 la sută din toate cazurile, boala apare în familii: cei afectați au moștenit mutația de la unul sau ambii părinți.
Până în prezent, au fost descrise mai multe mutații care cauzează sindromul Kallmann. Acestea includ mutațiile cu numele KAL1, FGFR1, FGF8, CHD7, SOX10, PROKR2 și PROK2.
Investigație și diagnostic
Diagnosticul sindromului Kallmann se face târziu în multe cazuri, deoarece simptomele devin adesea evidente doar în adolescență. Acest lucru se aplică în special băieților, la care lipsa dezvoltării sexuale este de obicei mai vizibilă. La fete, simptomele sunt adesea mai puțin pronunțate, astfel încât doar absența primei menstruații dă naștere la vizita unui medic. În plus, în multe cazuri, cei afectați nu suspectează o boală în spatele simptomelor, ci cred mai degrabă că se „dezvoltă târziu”.
În unele cazuri, boala este deja evidentă la sugari: băieții afectați pot avea testicule nedescendenți (criptorhidie) și / sau un penis foarte mic (micropenis).
Primul punct de contact dacă există semne de sindrom KS este medicul pediatru, în caz de lipsă de copil involuntară, ginecologul sau urologul.
Medicul efectuează următoarele examinări:
Antecedente familiale: Dacă se suspectează sindromul Kallmann, medicul întreabă dacă există cazuri cunoscute de SC în familie.
Examinarea fizică: caracteristicile sexuale secundare subdezvoltate, cum ar fi un penis prea mic pentru vârsta sa sau absența primei menstruații, oferă medicului primele indicii despre sindromul KS. De asemenea, acordă atenție mărimii corpului, axilei, pieptului și părului pubian și evaluează creșterea bărbii.
Test de miros: Acest test este posibil de la vârsta de aproximativ cinci ani. Medicul folosește parfumuri pure precum vanilina pentru a verifica dacă pacientul poate percepe mirosul.
Test de sânge: medicul va stabili un nivel hormonal modificat cu un test de sânge. De obicei, nivelurile de GnRH, LH și FSH sunt scăzute sau la un nivel normal scăzut. Nivelurile hormonilor sexuali la băieți și fete în adolescență sunt pre-pubescente.
Examinarea cu ultrasunete a testiculelor: medicul folosește ultrasunete pentru a examina țesuturile moi, cum ar fi testiculele.
Tomografie computerizată (CT) sau tomografie prin rezonanță magnetică (MRT): Cu aceste proceduri de examinare, medicul verifică dacă centrul olfactiv din creier (bulb olfactiv) este dezvoltat.
Examenul radiografic al mâinii: Cu ajutorul unei examinări radiografice a mâinii, medicul determină dacă plăcile de creștere sunt deja închise și dacă corpul a terminat astfel de creștere.
Spermiograma: medicul examinează dacă există ejaculare în sperma.
Test genetic: Testul genetic este utilizat pentru a determina mutația exactă care cauzează boala.
Cum este tratat sindromul Kallmann?
Sindromul Kallmann este ușor de tratat. Cei afectați primesc tratament de substituție cu hormoni sexuali. Dacă boala este recunoscută și tratată înainte de pubertate, cei afectați duc de obicei o viață în mare măsură nerestricționată. Aceasta include dezvoltarea regulată a pubertății și o viață sexuală normală.
Administrarea hormonului sexual: bărbații primesc testosteron, femeile estrogene și progesteron. Preparatele hormonale sunt disponibile sub formă de injecții, geluri sau plasturi. Terapia hormonală este continuată de obicei pe viață la bărbați și până la menopauză la femei.
În 10-20 la sută din cazuri, o deficiență congenitală de GnRH se rezolvă după terminarea terapiei de substituție hormonală. După terapie, pacienții au niveluri normale de hormoni și au maturitate sexuală normală. Din acest motiv, medicii recomandă întreruperea terapiei la fiecare 1-2 ani pentru a determina necesitatea continuării terapiei.
Oferind hormoni sexuali dacă doriți să aveți copii: organismul are nevoie de GnRH pentru a se forma sperma. Din acest motiv, bărbaților care doresc să creeze un copil li se administrează hormonul GnRH în loc de testosteron. Durează 18 - 24 de luni până când începe producția de spermă. În aproximativ 80 la sută din cazuri, bărbații sunt fertili după aceea. Bărbații cu testicule nedescendenți au un prognostic puțin mai favorabil.
Tratamentul osteoporozei: pacienții cu densitate osoasă redusă primesc calciu și vitamina D. În plus, sportul și exercițiile fizice sunt recomandate pentru întărirea oaselor.
Anosmie: În prezent, nu există terapie pentru a restabili simțul mirosului.
Psihoterapie: Pentru unii pacienți cu KS, boala este o povară psihologică gravă. Psihoterapeutul este contactul potrivit aici.
Evoluția bolii și prognosticul
Modul în care progresează sindromul Kallmann variază de la persoană la persoană. Simptomele variază de la pacient la pacient. Este posibil ca boala să fie deja observată în copilărie, diagnosticată în adolescență, sau deficitul hormonal să apară doar la vârsta adultă.
Dacă boala este diagnosticată în timp util, prognosticul este foarte bun. Maturizarea sexuală se realizează la toți pacienții cu tratament hormonal adecvat. Pacienții au o dezvoltare regulată a pubertății, sunt fertili și au o speranță de viață normală. Aproape toți pacienții care doresc să aibă copii devin fertili cu un tratament adecvat.
Dacă KS este recunoscut și tratat numai după vârsta de 16 ani, este posibil să fi crescut deja. Această modificare nu poate fi inversată nici cu medicamente.
Împiedica
Deoarece este o boală genetică, nu este posibilă nici o prevenire.
Etichete: îngrijire a pielii sarcina loc de muncă sănătos
















-kopfsache.jpg)